Open innovation is the Lifeline of Ono.
We are incorporating the latest technologies in our efforts to effectively develop innovative drugs, through proactive promotion of open innovation in drug discovery, utilizing AI.
Why Leverage AI for Drug Discovery?
The development of a new drugs requires more than 10 years and costs over 100 billion yen, with a success rate of just 0.004%. AI-driven drug discovery has the potential to provide breakthroughs in these extremely challenging situation. In addition to being able to analyze vast amounts of genetic data and chemical compound information in a short period of time, and find promising candidates, it allows us to identify candidates with a higher risk of failure and potential side effects. If human memory is “a single bookshelf,” AI is equivalent to “a huge library.” Through application of AI, we hope to significantly reduce the drug development time frame and improve our success rate.
* Source: Japan Pharmaceutical Manufacturers Association “JPMA Newsletter No. 224” p. 17

Ono’s AI Drug Discovery
For over 20 years, we have undertaken research leveraging technologies such as computational chemistry and simulation. Based on our strength in open innovation, we are also actively promoting co-creation with diverse partners, including pharmaceutical and hi-tech companies.
History of Digital- and AI-based Drug Discovery
| From around 1992– | Began centralized data management |
|---|---|
| 2017 | Began collaboration with Schrödinger, Inc., leveraging their cutting-edge simulation technology |
| From around 2018– | Began investigating use of generative AI |
| 2020 | Launch of Velexbru, developed in collaboration with Locus Pharmaceuticals, Inc. |
| 2021 | Began collaboration with Healx Ltd. |
| 2022 | Began collaboration with Iktos |
AI Drug Discovery
Utilizing Open Innovation
Participation in Tokyo-1 Project
Since 2023, Ono has been participating in a generative AI drug discovery project being advanced through the collaboration of four Japanese pharmaceutical companies. The project uses Tokyo-1, a large-scale GPU supercomputer, to provide an environment where high-performance computing resources are available for immediate use. We expect to dramatically accelerate the drug discovery research. It also provides an arrangement where pharmaceutical companies can exchange corporate information in fields where they are not competing. It is hoped that this sharing of knowledge through open innovation will contribute to enhancing the speed and quality of research.

The Front Line of Ono’s AI Drug Discovery
We are promoting the application of AI from the discovery of promising candidates in initial stages of drug discovery through to the stage when a candidate is selected for development as a pharmaceutical product, in an effort to improve the sophistication and efficiency of drug discovery.

Thorough analysis of vast amounts of research data through the power of AI enables us to detect seeds of drug discovery that remained hidden until now and to design compounds more efficiently, whereby we aim to significantly shorten development time and dramatically improve our success rate. But we are not just aiming for greater efficiency. We believe that the key to producing true innovation lies in striving for the optimal balance between that which should be entrusted to AI and the creativity and enthusiasm that only humans can exercise. By fusing AI with human wisdom, we will advance the very process of drug discovery endeavor. We will continue our efforts unceasingly, with strong enthusiasm and a sense of responsibility, to pave the way for a new tomorrow for all patients.
Hiromu Egashira
Senior Director, Drug Discovery Chemistry, Discovery & Research
Overall Image of Use of AI in Drug Discovery

Hit compound: A compound in early-stage drug discovery deemed active against target proteins.
Lead compound: A compound from among hit compounds that, through betterment of activity, selectivity, and safety, has improved characteristics as a drug candidate.
Drug candidate: A compound selected to proceed with pre-clinical trials. At this stage, it meets safety and efficacy standards and is considered a target for actual drug development.
1 Knowledge graphs
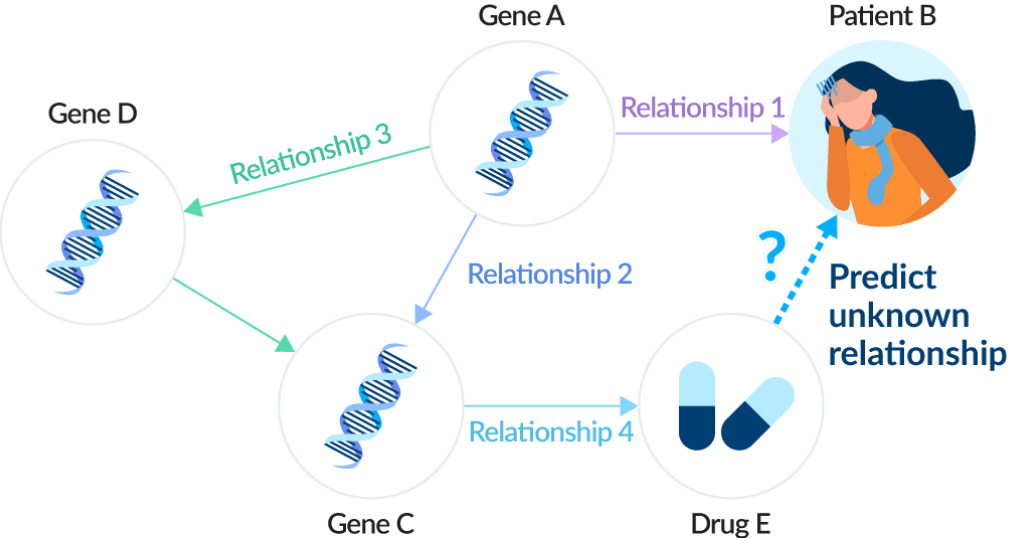
Ono has introduced advanced methods that combine AI and knowledge graphs aimed at forecasting and hypothesis generation in drug discovery. By structuring the relationship between genes, disease, and pharmaceuticals, we thereby extract useful knowledge. We use Tokyo-1 to handle extensive computation, achieving sophisticated analysis in a realistic time frame. Based on the results derived from collaboration with Healx and the use of open source, we are advancing construction of an independent model. In addition, we have introduced explainable artificial intelligence (XAI) and large-scale language models, working to integrate in-house data, with the aim of improving both the quality and speed of drug discovery.
2 Large-scale GPU docking
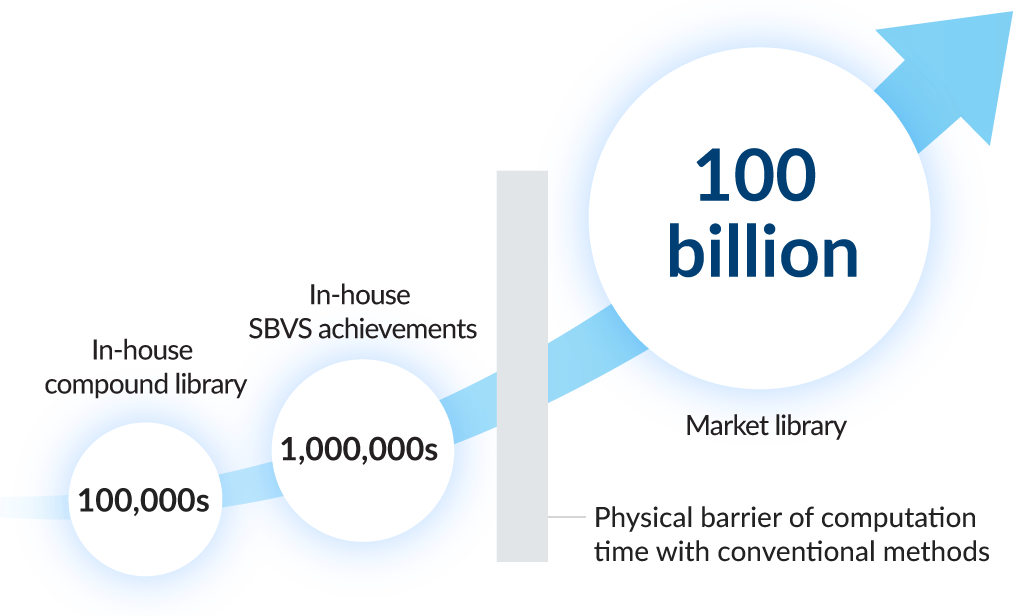
In the Tokyo-1 sub-working group, multiple pharmaceutical manufacturers are engaged in ultra-high-speed structure-based virtual screening (SBVS) using GPU. Through the development of small-molecule compound preliminary processing programs and high-speed tools, we are achieving processing speeds over 100 times faster than conventional methods. Each company has reported results, such as the complete analysis of seven million compounds in just a few days. At present, we are aiming to construct processes to enable screening at a challenging scale of 100 billion compounds in just days, in collaboration with AI.
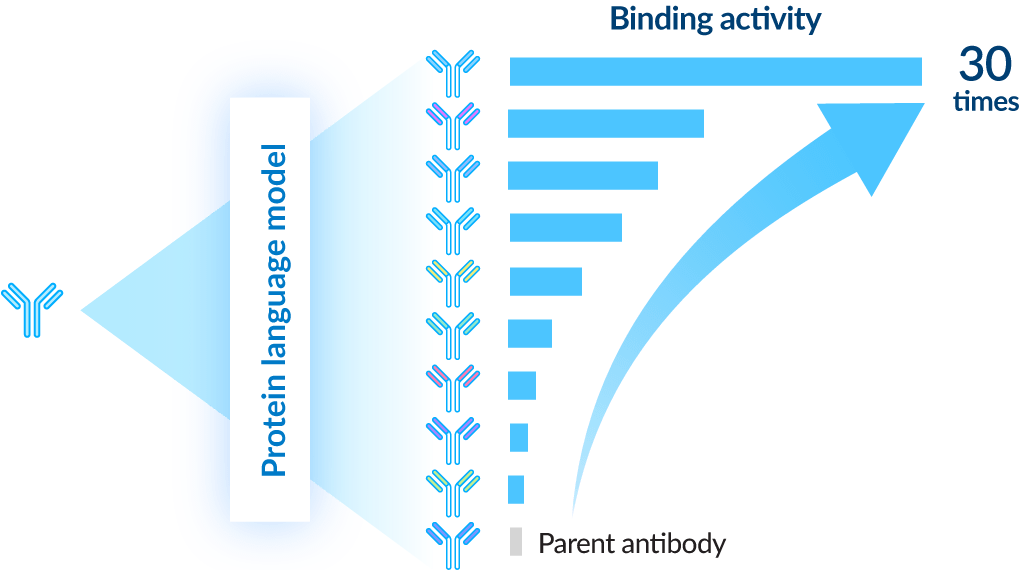
3 Protein language models
We are working to improve antibody parameter forecasting and activity using the various publicly available protein and antibody language models. By conducting extensive calculations using Tokyo-1, we identified antibodies with up to 30 times improved binding activity from among the tens of antibodies planned for testing. Currently, we are training foundational protein language models on antibody data with the goal of constructing a model capable of making more precise predictions.
performance to the parent antibody
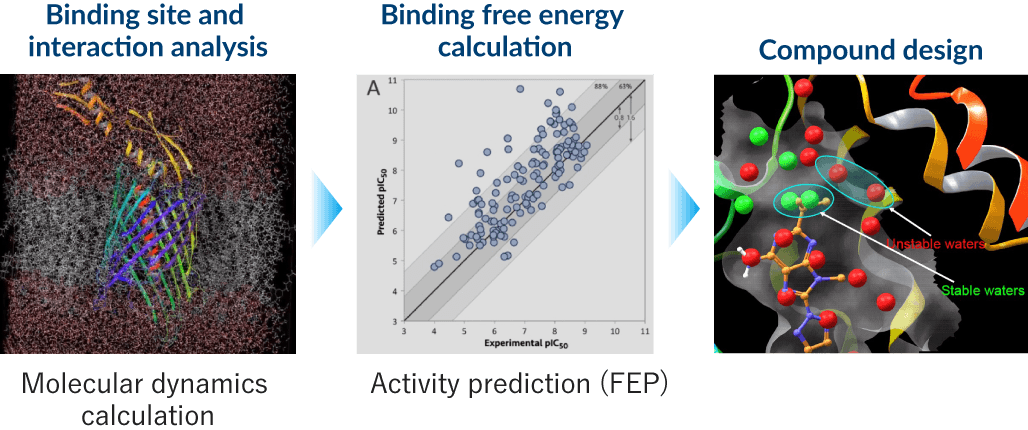
4 High-precision protein / low-molecular-weight compound binding forecasting
As part of our structure-based approach to drug discovery, we obtain three-dimensional protein structures using X-ray crystallography, cryo-electron microscopy, and AI technology. We conduct molecular dynamics calculations based on the structural information obtained, allowing for detailed analysis of the proteins and small-molecule compounds binding mode and interaction. In addition, we calculate the free energies of binding through free-energy perturbation plus (FEP+) for precision forecasting of compound binding strength. We use these results for the design of new compounds and optimization of candidates, striving for greater efficiency in drug discovery research.
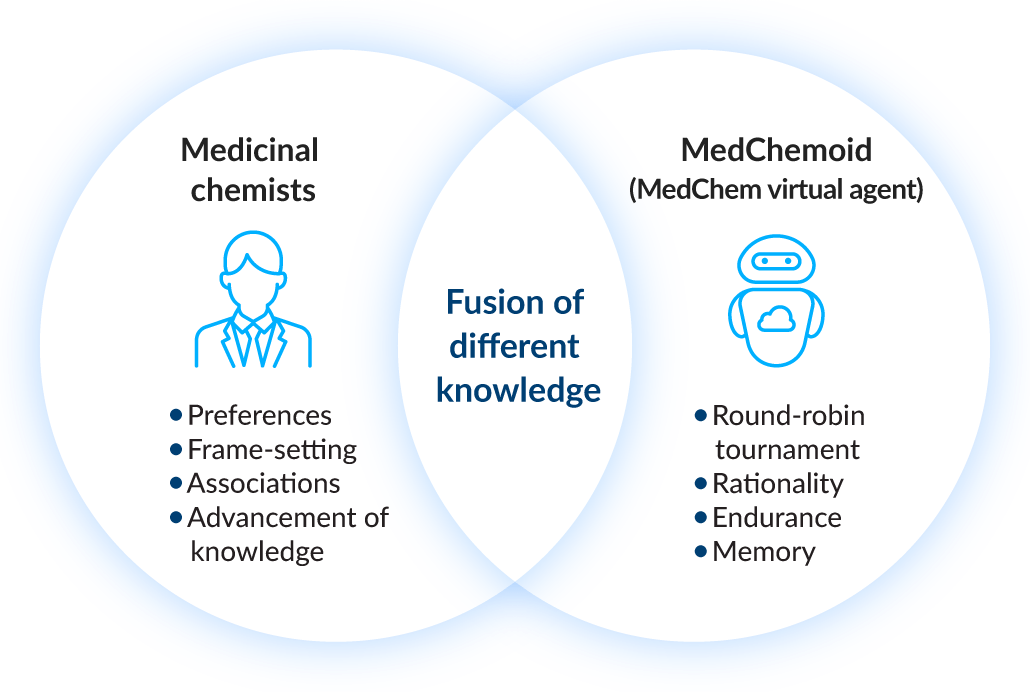
5 Co-existence of medicinal chemists and AI
We are driving “social drug discovery” through the collaboration of medicinal chemists and AI. By fusing the intelligence of medicinal chemists with that of AI, we aim to resolve issues in molecular design. Using the Tokyo-1 supercomputer, we are conducting reinforced learning of AI with feedback from medicinal chemists. Presently, we are proceeding with the development of a medicinal chemist AI agent and interaction tests between medicinal chemists and AI agents, accelerating implementation of a collaborative system whereby humans and AI advance together.
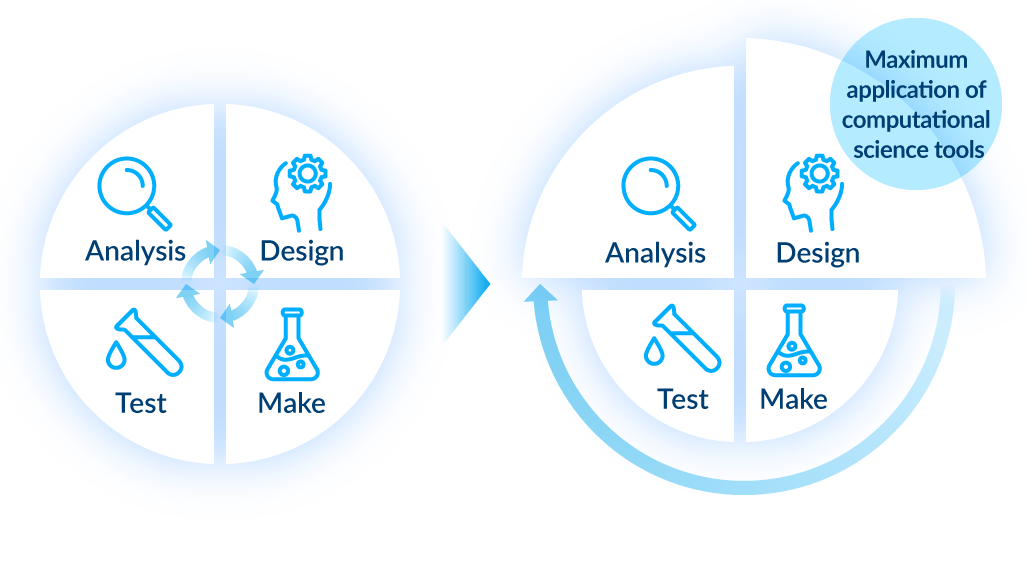
6 Compound optimization
We evaluate compound activity and physicochemical properties using visualized maps of compound design and synthesis. By utilizing AI forecasting to strengthen the design section of our Design - Make - Test - Analyze (DMTA) cycle in particular, we eliminate wasteful compositions in an effort to shorten the time frame. We use key evaluation data in the next stage of design and have introduced an automated synthesis machine for efficient synthesis. Through integration of computational science with experimental work, we aim to optimize the DMTA cycle and further accelerate drug discovery.